
多动症又称注意缺陷多动障碍(Attention Deficit Hyperactivity Disorder,ADHD),是中枢神经系统结构和功能异常的一种疾病,多见于儿童。以注意力不集中、多动、冲动为主要症状[1,2]。目前针对ADHD的药物治疗选择包括兴奋剂、非兴奋剂、抗抑郁剂、抗精神病药物等[3]。如表1所示,兴奋剂主要包括苯丙胺/右旋苯丙胺和哌醋甲酯,非兴奋剂包括托莫西汀、维洛沙嗪、胍法辛和可乐定等,发挥去甲肾上腺素(NE)再摄取抑制剂或α2受体激动剂的作用,其中托莫西汀和维洛沙嗪是NE再摄取抑制剂,胍法辛和可乐定则是α2受体激动剂[4]。此外,近年来美国食品和药物管理局(FAD)批准了一些复方制剂及剂型转换的药物,如维洛嗪缓释胶囊制剂(viloxazine ER;Qelbree®)、二甲磺酸赖右苯丙胺(Lisdexamfetamine dimesylate ,LDX)以及AZSTARYS (右哌甲酯(d-MPH)前药serdexmethylphenidate (SDX)和速释d-MPH组成的一种复方胶囊剂型,SDX/d-MPH)等。
表1 对小儿多动症发病机制具有作用的治疗药物
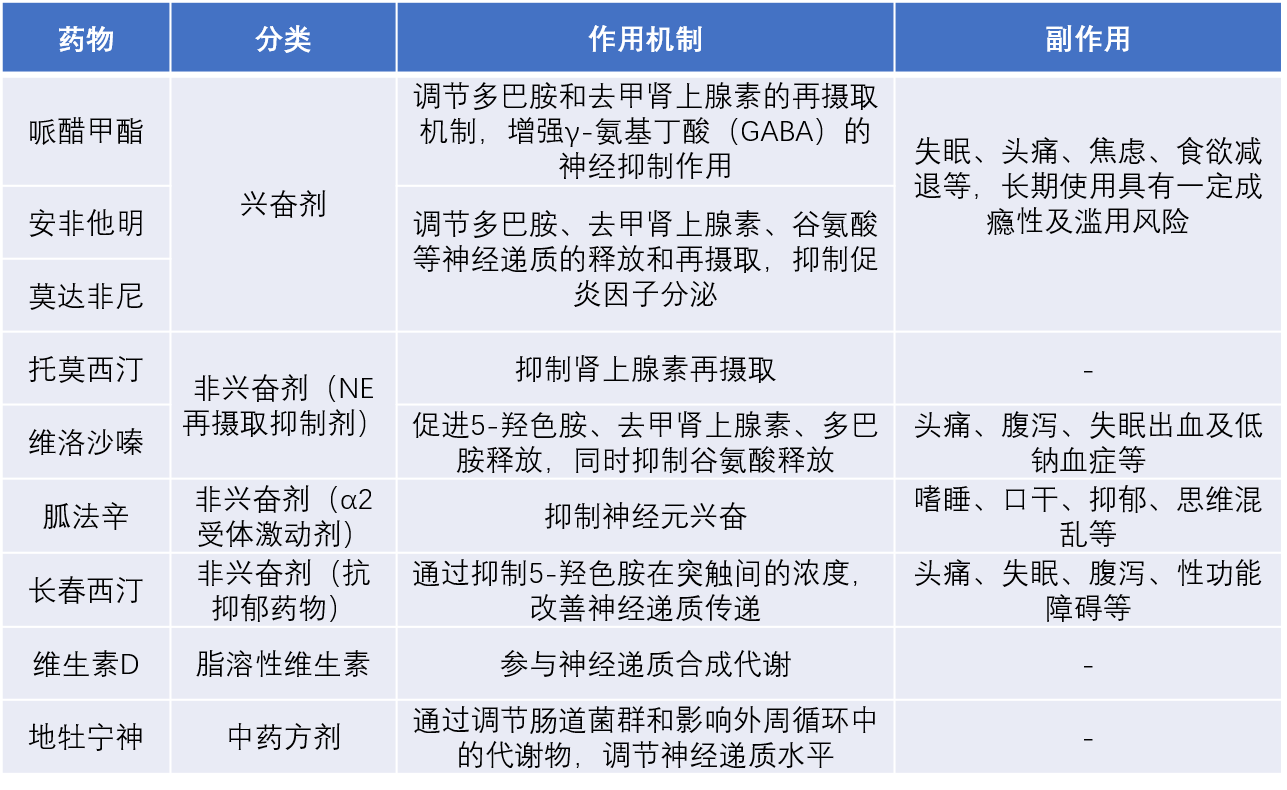
资料来源:作者整理
哌醋甲酯(Methylphenidate, MPH)是一种拟交感神经胺,可阻断多巴胺和去甲肾上腺素的再摄取机制,用于治疗ADHD[5]。脑源性神经营养因子/原肌球蛋白受体激酶B信号通路(BDNF/TrkB)在DA囊泡循环和ADHD发病机制中起关键作用,BDNF/TrkB信号通路可能通过影响与ADHD发病机制密切相关的蛋白受体复合物参与调节DA突触小泡循环,MPH可显著改善BDNF/TrkB信号通路的低激活状态,且能够显著增加PFC、纹状体和海马中BDNF/TrkB的表达[6]。有研究通过利用正电子发射断层扫描(PET)来研究MPH在人脑中的作用机制。证明了口服MPH在给药后60-90分钟在大脑中达到峰值浓度,治疗剂量的MPH阻断超过50%的DAT。并且,MPH可显著增强基底神经节中的细胞外DA,并受DA释放速率的调节[7]。
此外,与短效兴奋剂相比,缓释兴奋剂通常是首选,其优点在于作用时间持续、对多剂量的需求降低、具有更好耐受性的趋势以及减少滥用和转移的可能性[8]。美国FDA已经批准AZSTARYS的新药申请,用于治疗6岁及以上患者的注意力缺陷多动障碍。AZSTARYS中含有30%立即释放的d-MPH和70%延长释放的新型SDX,作为一种复方胶囊剂型,具有有起效快、减少药物依赖性的优点。
安非他明(Amphetamine, AM, 苯丙胺类)作为一种兴奋剂,通常用于儿童和成人ADHD的治疗,可作为速释、缓释和缓释口服药物使用[9]。安非他明表现出三种主要的作用机制。首先,它能够与单胺转运蛋白、NET和DAT结合,以防止各自神经递质的再摄取。其次,安非他明允许痕量胺相关受体1(TAAR1)磷酸化DAT,从而停止转运或逆转DA的流出。第三,安非他明可能进入突触前单胺囊泡,导致神经递质向突触流出[10]。安非他明作用包括DAT和NET抑制、囊泡单胺转运体2(VMAT-2)抑制和单胺氧化酶活性抑制[11]。此外,安非他明具有一定的成瘾性,高剂量的安非他明会损害血清素能和多巴胺能神经元,长期可能会出现持续的神经毒性脑损伤[12]。
二甲磺酸地塞米非他明(LDX)作为安非他明前药,已被美国FDA批准作为用于治疗6岁以上的儿童(当MPH治疗不理想时),也是治疗成人ADHD和暴食症的一线治疗药物[13]。LDX本身在药理学上是不活跃的,但口服后通过限速酶解转化为L-赖氨酸和D-苯丙胺。该药物的活性形式通过主要抑制DAT、NET、微量胺相关受体1(TAAR1)和囊泡单胺转运体2(SLC18A2)等靶点刺激中枢神经系统的活性,来调节突触间隙儿茶酚胺(主要是NE和DA)的再摄取和释放。给药后,LDX通过红细胞在血液中将L-赖氨酸裂解转化为活性药物D-苯丙胺[14]。
托莫西汀(Tomoxetine, ATX)是一种非中枢兴奋剂,是成人注意力缺陷/多动障碍(ADHD)的标准治疗方法[15]。托莫西汀能够抑制NET,导致前额叶皮层(Prefrontal cortex, PFC)突触间隙中NE和DA水平升高。值得注意的是,托莫西汀不会增加伏隔核(几乎没有NETs的大脑奖励区)中的NE或DA,因此,减少了滥用的可能性[16]。托莫西汀是突触前去甲肾上腺素转运蛋白的抑制剂,对多巴胺转运蛋白的亲和力非常低,去甲肾上腺素转运体主要负责清除前额叶皮层中的多巴胺;因此,托莫西汀会增加前额叶皮层的细胞外去甲肾上腺素和多巴胺,且能够降低背前扣带皮层和背外侧前额叶皮层的激活,这与注意力集中的改善相关[17]。
维洛沙嗪(Viloxazine)作为选择性5-HT 2B受体拮抗剂和5-HT 2C调节血清素活性受体激动剂,能够抑制NET,从而阻断NE的再摄取[18]。维洛沙嗪在不抑制血清素转运蛋白(SERT)的情况下表现出5-HT的增强作用,它对血清素受体5-HT 2C有一定的激动活性,对5-HT 2B有拮抗活性,对5-HT 7有弱拮抗活性。前额叶皮层是与ADHD相关的大脑区域,而维洛沙嗪可增加前额叶皮层的去甲肾上腺素、多巴胺和5-HT活性[19]。
Viloxazine缓释胶囊(Viloxazine ER)是一种新型非兴奋剂药物,已于 2021 年 4 月被美国FDA批准用于治疗儿童和青少年(6-17岁)的ADHD,具有治疗效果好及耐受性良好的特点[20]。药物III期试验表明,Viloxazine ER治疗使成人注意力缺陷多动障碍症状、执行功能和整体临床疾病严重程度有所改善。同时,因不良事件停药率仅为9.0%,也表明Viloxazine ER具有良好的耐受性[21]。
长春西汀(Vinpocetine)是一种常用的磷酸二酯酶抑制药,可防止兴奋性氨基酸所致的受体过度兴奋,减少脑细胞兴奋中毒性死亡;提高脑组织对氧气及葡萄糖的摄取、利用效率,增加能量供应,促进5-羟色胺、多巴胺等神经递质释放,减轻焦虑、抑郁等情绪[22]。长春西汀可有效疏通血管、抑制血小板聚集、降低血液黏滞性,同时也可抑制单核细胞黏附、趋化,有效减少炎症细胞浸润,充分发挥自身抗炎作用,减轻神经功能障碍,因此长春西汀能够使炎症因子水平有明显降低,减少脑组织损伤[23]。
肠道微生物群可能会干扰儿茶酚胺能神经传递系统,通过影响其代谢途径或神经递质转运蛋白的基因表达而导致ADHD[24]。除了神经兴奋剂之外,许多草药或草药化合物对不同中枢神经系统疾病具有治疗作用,揭示了肠道微生物群的变化在药物的药理作用中起关键作用[25]。中药复方口服制剂地牡宁神(Dimuningshen, DMNS)已广泛应用于ADHD的临床治疗,DMNS主要通过调节肠道菌群和影响外周循环中的代谢物,对ADHD产生治疗作用。
ADHD是一种具有强烈遗传基础的神经精神疾病,ADHD病因学中存在遗传背景和环境因素之间的相互作用[26]。它的高患病率以及对其治疗的持久争议,推动了科学和社会对其病因和机制的研究兴趣。ADHD具有一定的持续性,以及共病率。有研究[27]发现,虽然持续性成人(儿童期发病)ADHD患病率为2.58%,症状性成人ADHD(无儿童期发病)患病率为6.76%,在人口结构中有一定占比率。而ADHD共病则包括自闭症、抽动障碍、癫痫、抑郁以及肥胖等。ADHD的发病机制除NE、DA、神经炎症和肠道菌群外,缺钙、微量元素、锌等也可能是引发ADHD的原因。目前,治疗ADHD的针对性药物如哌醋甲酯等中枢兴奋剂具有较多的药物反应,能够诱发和加重共病症状,且存在药物滥用的问题,争议较大。ADHD病因复杂,目前仍不明确其发病机制,故明确ADHD的作用机制,寻找治疗ADHD的药物势在必行。
体内模型
幼龄大鼠自发性多动症模型
母鼠孕期暴露尼古丁诱导的仔鼠多动症模型
检测指标
旷场试验
高架十字迷宫试验
新物体熟悉与识别试验
前额叶及纹状体DA、NE、cAMP表达水平
前额叶、纹状体α2、D1、D2受体表达水平

微透析技术研究大脑递质分泌
前额叶及纹状体DA、NE及其代谢物的分泌
参考文献


